Introduction to Thermal Analysis
The essence of thermal analysis is temperature analysis.
Thermal analysis technology is to measure the change of physical properties of substances with temperature under the control of programmed temperature (i.e. constant rate of temperature rise, constant rate of temperature drop, constant temperature or step temperature rise, etc.), which is used to study the change of physical parameters such as thermal, mechanical, acoustic, optical, electrical, magnetic, etc. of substances at a specific temperature, namely P=f (T).
The temperature change is designed according to a certain rule, that is, the program control temperature: T=(t), so its property is both a function of temperature and time: P=f (T, t).
Significance of material thermal analysis
It is widely used to characterize the thermal, physical, and mechanical properties and stability of materials.
It is of great practical significance for the research and development of materials and the quality control in production.
Review of the brief history of thermal analysis
Interpretation of Common Thermal Analysis Methods
According to the induction and classification of the International Thermal Analysis Association (ICTA), the current thermal analysis methods are divided into nine categories and seventeen kinds.
The commonly used thermal analysis methods include thermogravimetric analysis (TG), differential scanning calorimetry (DSC), static thermomechanical analysis (TMA), dynamic thermomechanical analysis (DMTA), dynamic dielectric analysis (DETA), etc.
They are functions of measuring material weight, heat, size, modulus, compliance, dielectric constant and other parameters on temperature.
| Physical property | Analysis technology name | abbreviation | physical property | Analysis technology name | abbreviation |
| 1. Quality | 1) Thermogravimetry | TG | 3. Enthalpy | 9) Differential scanning calorimetry | DSC |
| 2) Isobaric mass change measurement | 4. Dimensions | 10) Thermal expansion method | |||
| 3) Detection of escaping gas | EGD | 5. Mechanical properties | 11) Thermomechanical analysis | TMA | |
| 4) Escape gas analysis | EGA | 12) Dynamic Thermomechanical Analysis | DMA | ||
| 5) Radiothermal analysis | 6. Acoustic characteristics | 13) Thermoacoustic method | |||
| 6) Thermal particle analysis | 14) Thermoacoustic method | ||||
| 2. Temperature | 7) Determination of heating curve | 7. Optical characteristics | 15) Thermooptic method | ||
| 8) Differential thermal analysis | DTA | 8. Electrical characteristics | 16) Thermoelectricity method | ||
| 9. Magnetic characteristics | 17) Thermomagnetic method |
(1) Thermogravimetric analysis (TG)
Thermogravimetry (TG) is a technique to measure the change of sample mass with temperature or time under the control of programmed temperature.

Scope of application:
(1) The chemical changes such as thermal stability, thermal decomposition and oxidative degradation of materials in inert gas, air and oxygen are mainly studied;
(2) All physical processes involving mass change are studied, such as determination of moisture, volatile matter and residue, absorption and desorption, gasification rate and gasification heat, sublimation rate and sublimation heat, the composition of polymer or blend with filler, etc.
Principle explanation:
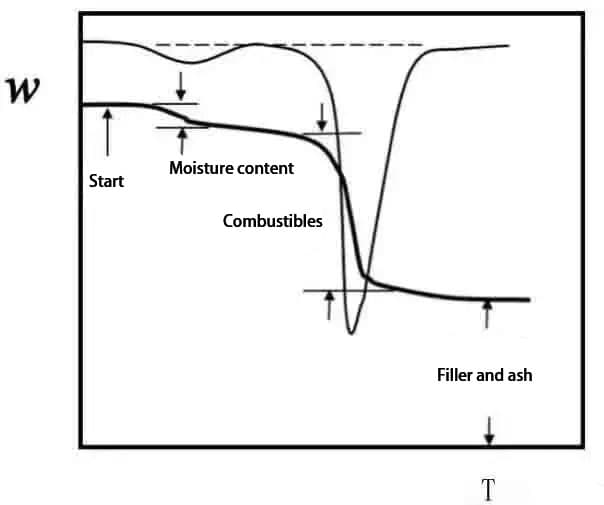
The thermogravimetric curve (TG curve) is obtained by plotting the sample weight fraction w against the temperature T or time t: w=f (T or t).
Because most of the temperature rise is linear, T and t are only one constant different.
The first derivative of TG curve with respect to temperature or time, dw/dT or dw/dt, is called differential thermogravimetric curve (DTG curve).

In Fig. 2, the cumulative weight change at Ti at point B reaches the lower limit of thermobalance detection, which is called the initial reaction temperature;
The change of weight at point C Tf can not be detected, which is called the end of reaction temperature;
Ti or Tf can also be determined by extrapolation, which is divided into G point and H point;
The temperature when the weight loss reaches a predetermined value (5%, 10%, etc.) can also be taken as Ti.
Tp represents the maximum weight loss rate temperature, corresponding to the peak temperature of DTG curve.
The peak area is proportional to the weight change of the sample.
Practical application:
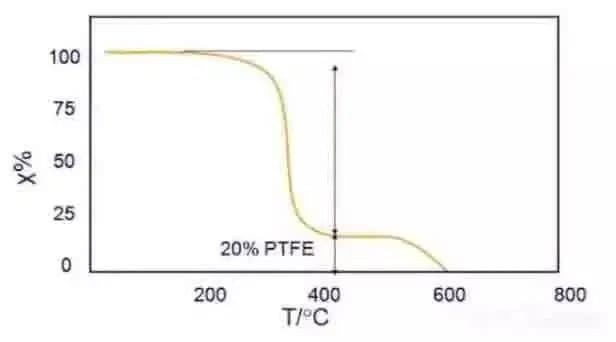
Thermogravimetry has become an important method to study the thermal change process of polymers because of its rapidity and simplicity.
For example, the TG curve of the blend of PTFE and acetal copolymer in Fig. 3 can be used to analyze the components of the blend.
It can be found from the figure that when heated in N2, the acetal component decomposes (about 80%) at 300~350 ℃, and the PTFE starts to decompose (about 20%) at 550 ℃.
Influencing factors:
(a) Heating rate:
The faster the temperature rises, the greater the temperature lag, the higher the Ti and Tf, and the wider the reaction temperature range.
It is suggested that polymer sample should be 10K/min, inorganic and metal sample should be 10-20K/min;
(b) Particle size and dosage of sample:
The particle size of the sample should not be too large and the compactness of the filling should be moderate.
For the same batch of test samples, the particle size and packing tightness of each sample shall be consistent;
(c) Atmosphere:
Common atmospheres include air, O2, N2, He, H2, CO2, Cl2 and water vapor.
The reaction mechanism is different in different atmospheres. When the atmosphere reacts with the sample, the shape of TG curve is affected;
(d) The material and shape of the sample dish.
(2) Static Thermomechanical Analysis (TMA)
Thermomechanical analysis is a technique to measure the functional relationship between the deformation of materials and temperature time under the action of program temperature and non vibration load, mainly measuring the expansion coefficient and phase transition temperature of materials.
Scope of application:
- Static thermomechanical analyzer is mainly used for thermal expansion coefficient of inorganic materials, metal materials, composite materials and polymer materials (plastics, rubber, etc.);
- Glass transition temperature;
- Melting point;
- Softening point;
- Load thermal deformation temperature;
- Creep, etc.
Practical application:
(a) Research on fiber and film:
It can measure its elongation, shrinkage performance, modulus and corresponding temperature, stress strain analysis, and stress analysis under freezing and heating conditions;
(b) Characterization of composite materials:
In addition to the study of fiber with TMA, the reinforcement of composite materials, the glass transition temperature Tg, gel time and fluidity of resin, thermal expansion coefficient and other properties, as well as the dimensional stability and high-temperature stability of multilayer composite materials can be quickly measured and studied with TMA;
(c) Research on coatings:
It is possible to know whether the coating matches the substrate and the matching temperature range;
(d) Research on rubber:
It can be known whether the rubber still has elasticity and whether the size is stable in harsh use environment.
Influencing factors:
(a) Heating rate:
The temperature distribution of the sample is uneven when the heating rate is too fast;
(b) Sample thermal history;
(c) Sample defects:
Porosity, uneven distribution of filler, cracking, etc;
(d) Pressure applied by probe:
0.001~0.1N is generally recommended;
(e) Chemical change of sample;
(f) External vibration;
(g) Calibration:
Calibration of probe, temperature, pressure, furnace constant, etc;
(h) Atmosphere;
(i) Sample shape
Whether the upper and lower surfaces are applied in parallel.
(3) Differential scanning calorimetry (DSC)

Principle explanation:
Differential scanning calorimetry (DSC) is a technology to measure the relationship between the power difference between the material and the reference material and the temperature under the program control temperature.
There are two kinds of differential scanning calorimetry: compensation type and heat flow type.
Two groups of compensating heating wires are installed under the sample and reference object containers.
When the temperature difference ΔT between the sample and the reference object occurs due to the thermal effect during the heating process, the current flowing into the compensating heating wire will change through the differential thermal amplifier circuit and differential thermal compensation amplifier.
When the sample absorbs heat, the compensation amplifier will immediately increase the current on one side of the sample;
On the contrary, when the sample is exothermic, the current on one side of the reference material will increase until the heat on both sides is balanced and the temperature difference ΔT disappears.

In differential scanning calorimetry, the relation curve between the heat applied and the temperature required to keep the temperature difference between the sample and the reference at zero per unit time is a DSC curve.
The vertical axis of the curve is the amount of heating per unit time, and the horizontal axis is the temperature or time.
The area of the curve is proportional to the change of enthalpy. A typical DSC curve is shown in Fig. 4.
Scope of application:
(1) Determination of curing reaction temperature and thermal effect of materials, such as reaction heat, reaction rate, etc;
(2) Determination of thermodynamic and kinetic parameters of substances, such as specific heat capacity, heat of transformation, etc;
(3) Determination of crystallization, melting temperature and thermal effect of materials;
(4) Purity of the sample, etc.
Influencing factors:
(a) Heating rate:
The actual test results show that too high heating rate will cause uneven temperature distribution in the sample, and the furnace body and the sample will also produce thermal imbalance, so the influence of heating rate is very complex.
(b) Atmosphere:
Different gases have different thermal conductivity, which will affect the thermal resistance between the furnace wall and the sample, and affect the peak temperature and enthalpy.
(c) Sample dosage:
Not too much, so as to avoid expansion of peak shape and reduction of resolution due to slow internal heat transfer and large temperature gradient.
(d) Particle size of sample:
When the particle size of powder is different, due to the influence of heat transfer and diffusion, there will be differences in test results.
(4) Dynamic Thermomechanical Analysis (DMA)
Dynamic thermomechanical analysis measures the relationship between the mechanical properties of viscoelastic materials and time, temperature or frequency.
The sample is deformed under the action and control of periodic (sinusoidal) mechanical stress.
Scope of application:
The dynamic thermomechanical analyzer is mainly used to test the glass transition temperature, load thermal deformation temperature, creep, storage modulus (rigidity), loss modulus (damping performance), stress relaxation, etc. of inorganic materials, metal materials, composite materials and polymer materials (plastics, rubber, etc.).
DMA basic principle:
DMA characterizes the properties of materials through the state of molecular motion.
The molecular motion and physical state determine the dynamic modulus (stiffness) and damping (the energy lost by the sample in vibration).
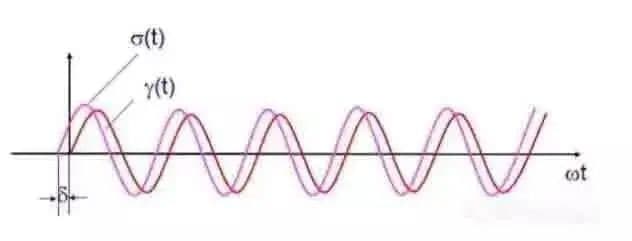
When a variable amplitude sinusoidal alternating stress is applied to the sample, a sinusoidal strain of preselected amplitude will be generated, and the strain on viscoelastic samples will lag behind a certain phase angle δ, as shown in Fig. 5.
DMA technology divides the viscoelasticity of materials into two moduli:
Storage modulus E’:
E’ is in direct proportion to the maximum elasticity of the sample stored in each week, reflecting the elastic components in the viscoelasticity of the material and characterizing the stiffness of the material;
The loss modulus E “:
E” is in direct proportion to the energy consumed by the sample in the form of heat in each week, reflecting the viscous part of the viscoelasticity of the material and representing the damping of the material.
The damping of the material also becomes the internal friction, expressed as tanδ, and the ratio of the energy lost by the material in the weekly period to the maximum elastic storage energy is equal to the loss modulus E “and the storage modulus E ‘of the material.

DMA adopts temperature rise scanning, from the auxiliary ambient temperature to the melting temperature, tanδ shows a series of peaks, and each peak will correspond to a specific relaxation process.
The phase angle tanδ, loss modulus E “and storage modulus E ‘can be measured by DMA as a function of temperature, frequency or time.
It not only gives mechanical properties in a wide range of temperature and frequency, but also can detect the glass transition, low temperature transition and secondary relaxation process of materials.
For example, the loss peak can represent the transition of certain unit motion.
Fig. 6 shows the curve of polystyrene tg changing with temperature, from which it can be inferred that the peak may be the movement of phenyl around the main chain;
The peak is the movement of benzene around the bond connecting the main chain.
Influencing factors: heating rate, sample thickness, presence or absence of metal coating, fixture type, etc.
(5) Dynamic dielectric analysis (DETA)
Dynamic dielectric analysis is a technology to test the change of dielectric properties of materials with temperature when materials are heated by a certain controlled temperature program under an alternating electric field of a certain frequency.
Dielectric analysis principle:
The dielectrics with dipoles will be directionally arranged with the external electric field under the action of the external electric field.
The polarization of the dipole is related to temperature and is accompanied by energy consumption.
Generally, the dielectric constant (ε) represents the degree of polarization of the dielectric under the external electric field, while the dielectric loss (D) represents the energy loss caused by polarization heating under the external electric field.
The directional arrangement of the dipoles under the action of the external electric field will also recover to the disordered state with the removal of the external electric field.
The time required for the dipole to recover from the regular arrangement to the random arrangement is called “dielectric relaxation time T”, according to Debye’s theory:

η is the medium viscosity, a is the molecular radius, K is the Boltzmann constant, and T is the temperature K.
The relaxation time is related to the size and shape of molecules and the viscosity of the medium. And

Where: tgδ is the tangent of loss angle, and ε0 is the dielectric constant under electrostatic field; ε∞ is the dielectric constant at the optical frequency.
It can be seen that ε and tgδ are physical quantities related to relaxation time τ, and therefore related to molecular structure, size and medium viscosity, which is the basis for using dielectric properties to study the molecular structure of substances.
It can be proved from the above two equations that when:

When, ε ‘has a maximum value, f0 is called “polarization frequency”.

That is, when the frequency of the external electric field is the polarization frequency, the dielectric loss is very large.
Scope of application:
This technique has been widely used to study the molecular structure, polymerization degree and polymer mechanism of dielectric materials.
In terms of application objects, there are thermoplastic and thermosetting resins such as polyacrylate methyl ester, polyvinyl chloride, polyamide, polyimide, polystyrene, phenol formaldehyde, epoxy, and wax.
In addition, there are polyphenyl maple and polybenzimidazole in high-temperature resistant resin, and proteins in biological compounds.
Its specific applications also include reinforced plastics, molding materials, coatings, adhesives, rubber, glass, ceramics and other metal oxides.
In the laboratory, DETA can be used as a powerful tool for viscoelastic research, such as dynamic mechanical properties and thermal mechanical properties testing.
In industrial production, it can be used in resin manufacturing, quality control, pre curing and curing degree control.


